Integrative structures of protein assemblies
An integrative structure determination approach combines diverse experimental data with physical principles, statistics of previous structures, and prior models for structure determination, making it a powerful method for studying macromolecular assemblies that are challenging to resolve using any single experimental method.
Over the past few years, several advancements in computational and experimental methods have sparked renewed enthusiasm for integrative modeling. AI-based structure prediction methods, such as AlphaFold, have transformed protein structure prediction. Whereas rapid advances in experimental techniques such as cryo-electron tomography (cryo-ET) now enable structural characterization of assemblies in their native cellular context at higher throughput and larger scales than before. Our research follows two synergistic tracks.
Integrative structure determination applied to biological systems
One track involves determining integrative structures of specific biological systems to gain insights into their function through close collaborations with experimentalists. We determined the structures of chromatin-associated assemblies, such as the nucleosome remodeling and deacetylase complex, and cell-cell junctions, such as the epithelial desmosomal outer dense plaque, combining diverse biophysical and biochemical data at various scales. Together, these integrative structures reveal mechanisms by which these complex molecular machines function and assemble; they also enabled us to rationalize mutations from cancers, epithelial, and cardiac diseases.
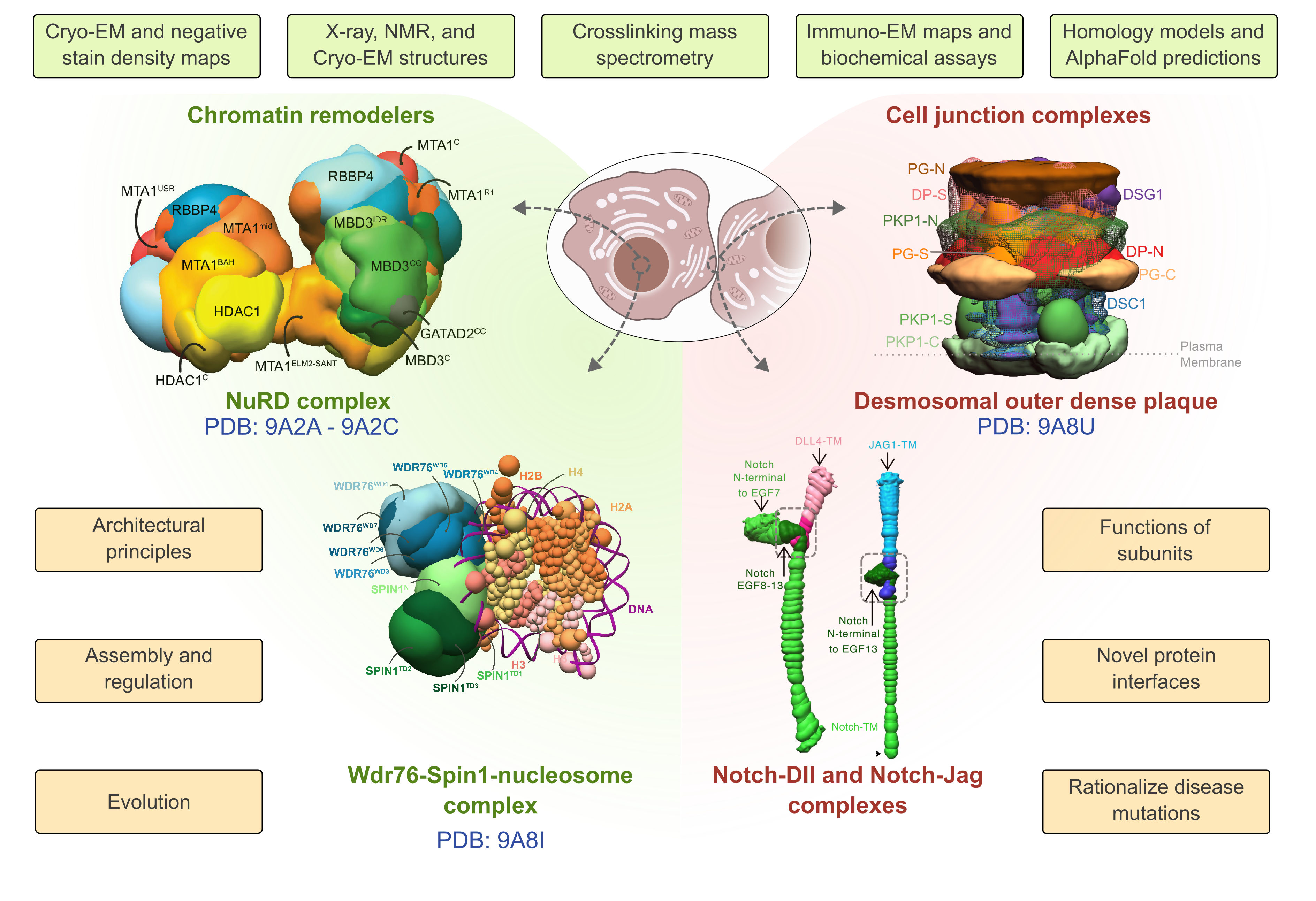
Integrative structures characterized recently are shown, along with the information that was incorporated (green background), and the insights derived (orange background).
Method development in integrative structural biology
A second research track involves developing broadly applicable, cutting-edge modeling methods, using algorithms from statistical inference, machine learning, computer vision, and related fields. We develop rigorous methods and software to make integrative structure determination more accurate and efficient by improving upon approaches that are ad hoc, semi-automated, based on trial-and-error, and/or require manual expert intervention. All our methods are available as open-source software. (See Software & Resources).
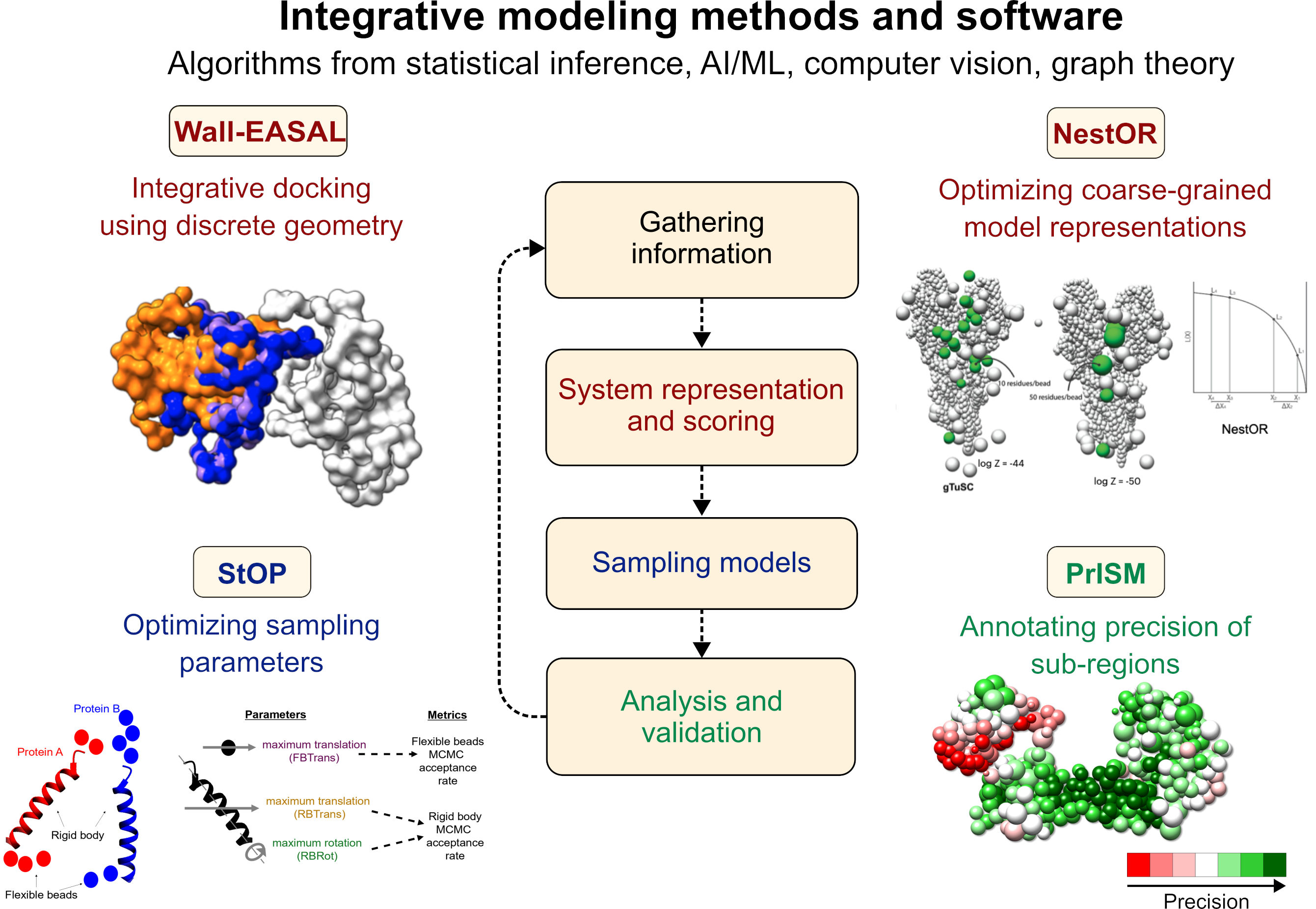
Integrative modeling methods and software developed by our group are shown. The text color indicates the corresponding stage of integrative modeling (center).
Usually, these method development directions emerge from recurrent themes identified across multiple modeling studies. Recent examples include Disobind, a deep learning method for identifying binding sites of intrinsically disordered regions (IDRs), and PickET, an unsupervised method for localizing macromolecules in cryo-ET data.

Disobind: a deep learning method for predicting interface residues and inter-protein contact maps for an IDR and its partner, given their sequences. Method architecture (top) and representative examples (bottom) are shown.
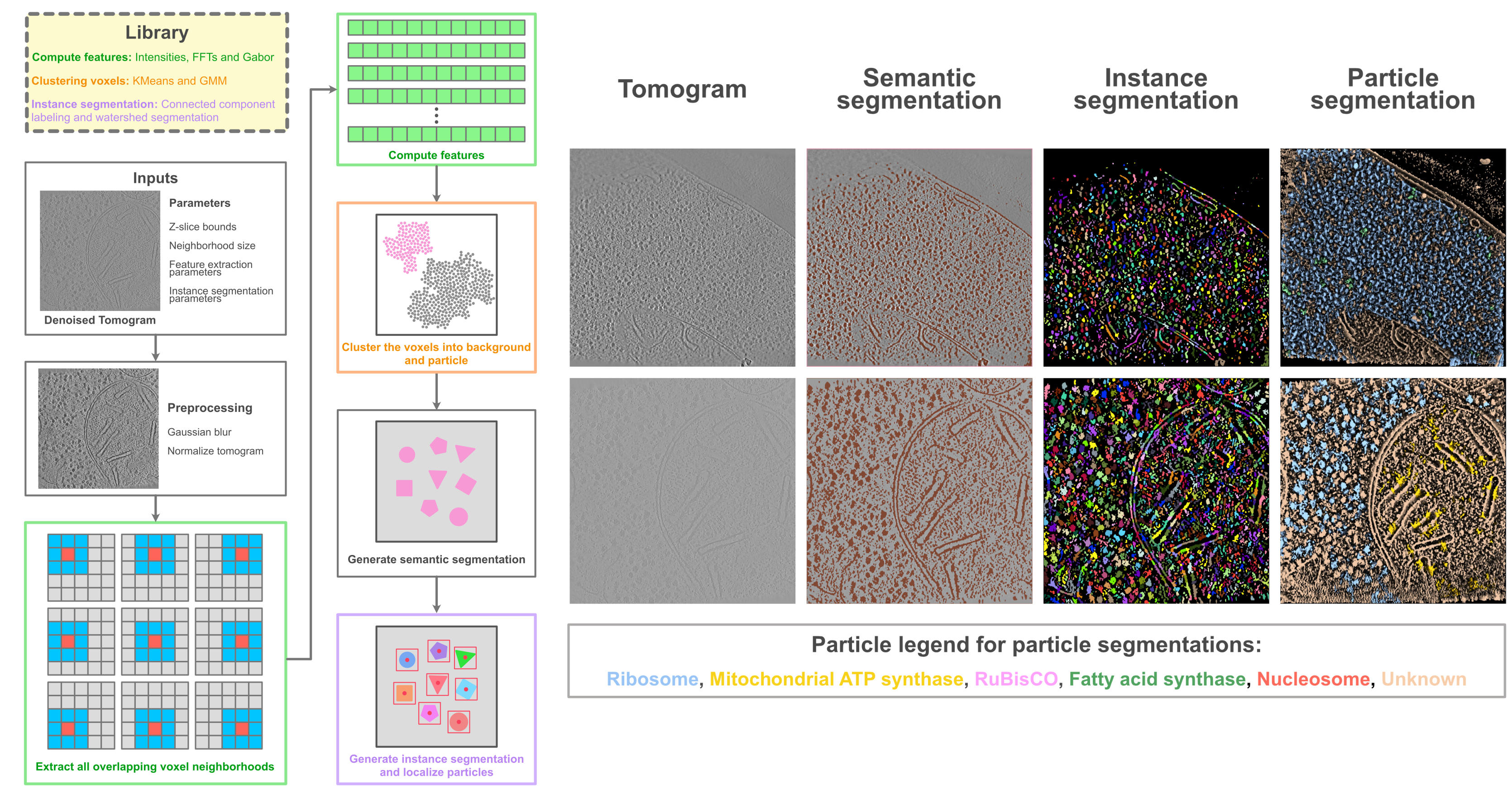
PickET: a workflow for unsupervised localization in tomograms (left) along with representative segmentations (right) for two real tomograms (CZI-DS-10001, gallium FIB milled S. pombe lamella: top row, CZI-DS-10301, plasma FIB milled C. reinhardtii lamella: bottom row). See PickET for more details.
We thank the following journals for highlighting our integrative modeling studies in their cover.

